
Integrative genomic analyses for newborn screening
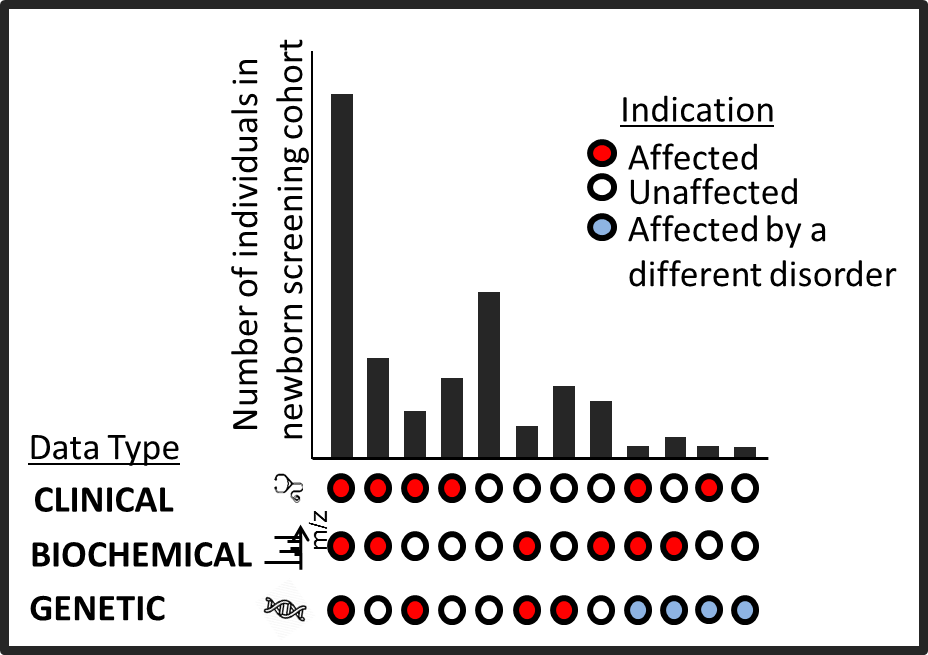
Newborn screening identifies newborns with disorders that are rare but treatable if caught early. It is currently performed using tandem mass spectrometry (MS/MS) which flags elevated metabolites in the blood. For some disorders, however, MS/MS is insensitive and imprecise. Therefore, we are exploring if genome sequencing can augment or replace MS/MS for newborn screening. In a collaboration with the California Department of Public Health, we sequenced dried blood spots from all California newborns born between 2005 to 2013 and diagnosed with a rare newborn screening disorder. Our results were surprising - sequencing analysis missed several more disorder cases compared to MS/MS, but also provided confident clinical resolutions in some particular cases where MS/MS followup required complex differential diagnoses. The findings have been presented and featured in multiple venues ( ASHG 2016 plenary talk, Science , GenomeWeb). Currently, we are integrating the mutational data from the genomes with the biochemical data form MS/MS as well as the follow-up clinical data to enhance newborn screening.
Rare disease diagnosis
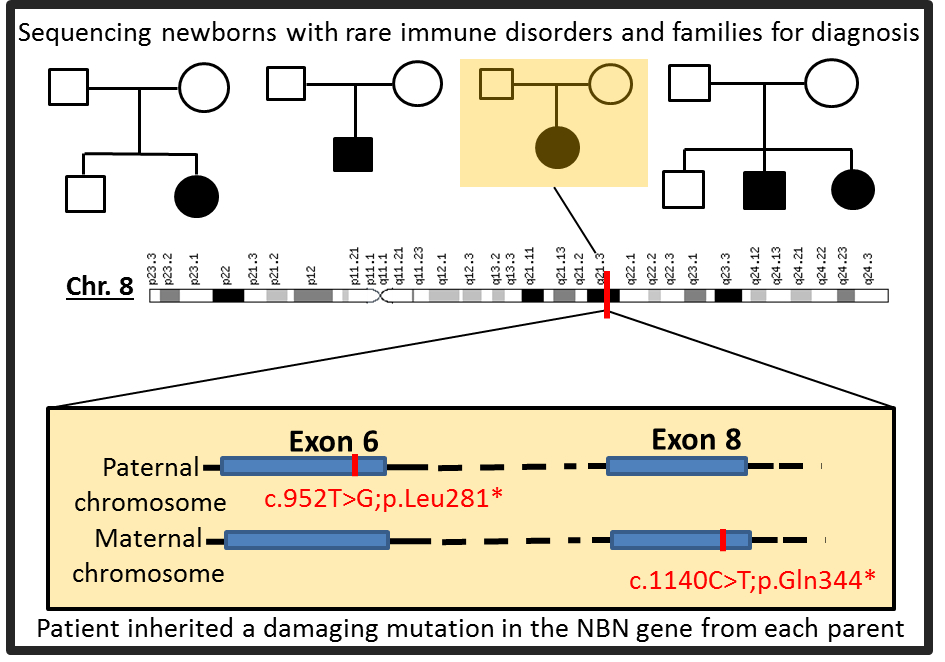
I analyze genomes of newborns with rare, undiagnosed disorders to resolve the genetic basis of their disorders. Many were identified from population based newborn screening for severe combined immundeficiencies (SCID), that revealed low or absent T cell receptor excision circles (TRECs), a biomarker for T-cell lymphocyte production. To interpret genomic variant data, we developed an analysis protocol whose distinctive features enabled solving numerous clinical cases. It integrates variant annotation, variant filtering, and gene prioritization to prioritize millions of called variants to a manageable shortlist of possible causative variants. We are able to make a definitive diagnosis of several rare immune related disorders with uncharacteristic symptoms with success rate of ~50% in cases with parent-child trios. Early detection provides information to offer prompt appropriate treatment and guidance, family genetic counseling, and avoidance of the diagnostic odyssey.
From mutations to mechanisms
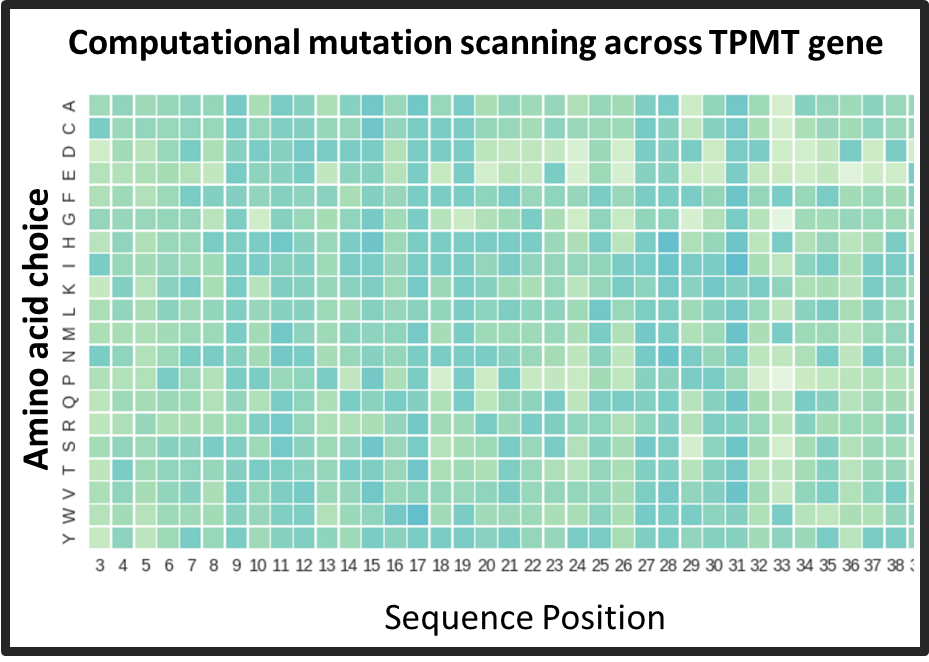
We are not yet as good at interpreting the genomic data as we are at generating them. Still, identifying the disease-causing mutations in the genome is merely the first step in untangling the downstream effects that lead to disease. Biological systems are characterized by complex and dense network of interactions among the molecules in a cell. Even in the simplest monogenic genetic disorders, our understanding of disease etiologies is rudimentary. Using phenylketonuria as a model monogenic disorder, I am studying how interactions among different mutations in the PAH gene can modulate disease severity. Analysis of large global genomic datasets of PKU patients has revealed interesting mutational relationships which are often population-specific. I am also working on computational methods to integrate high throughput biophysics measurements of intermediate phenotypes like protein stability and enzyme activity to produce better genetype-phenotype relationships in monogenic disorders.
Protein physics fundamentals
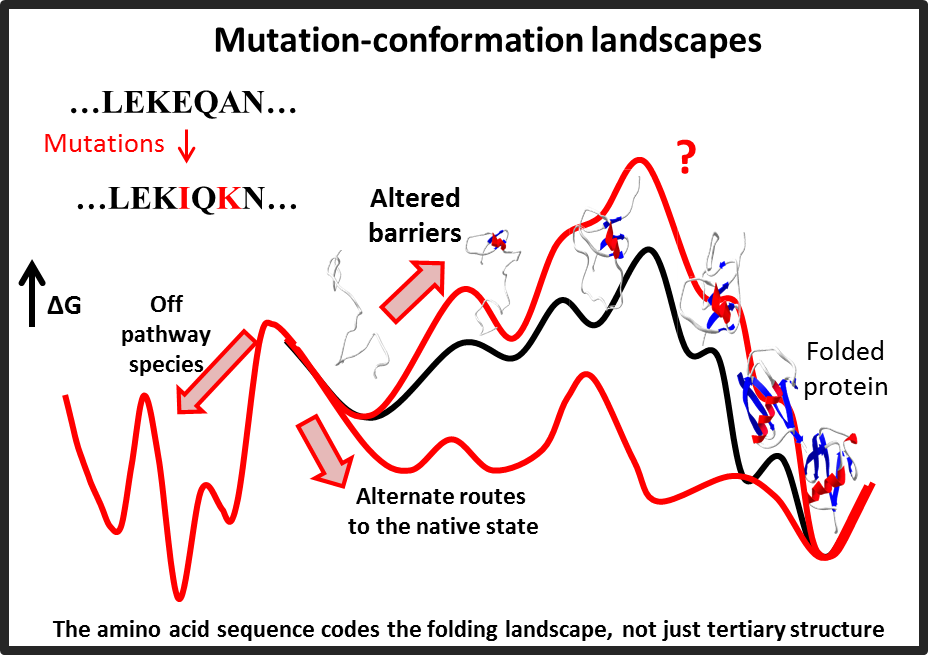
I have a longstanding interest in understanding how the fundamental interactions in biomolecules give rise to their structure and dynamics. I have developed computational models for protein folding that unify the prediction of a protein's folding pathways as well as secondary/tertiary structure using only is primary amino acid sequence. The underlying algorithm uses a search strategy inspired by the observation that proteins in nature fold in a sequential fashion by incrementally stabilizing native-like sub-structures or "foldons". I have adapted similar ideas towards several protein engineering applications. I am currently exploring methods to understand the effect of mutations in protein conformational landscapes.