|
Main Menu |
|||
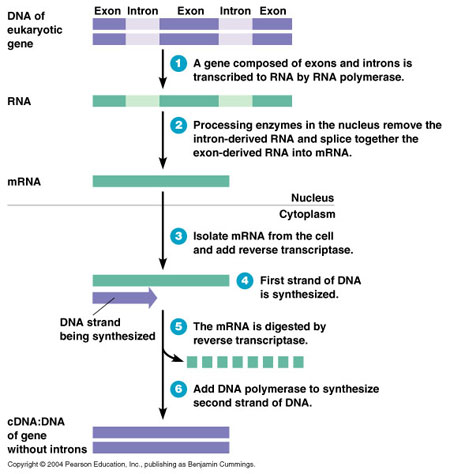
Computational GenomicsAs was previously mentioned, an organism's genome contains a lot of repeating, noncoding regions of DNA in addition to the useful sequences that encode proteins. Thus, because only the coding regions encode proteins, it is useful to look at those, as they are the sequences that will have an effect on the physiology of the cell. In addition, even in the coding region of most organisms, there are sequences that are transcribed, but not translated into a functional protein called Introns. Recall that DNA is transcribed by RNA Polymerase into an mRNA transcript. Before leaving the nucleus, however, this mRNA transcript often goes through splicing to remove any Introns that exist in the mRNA sequence before being translated into a protein by Ribosomes. Therefore, it is difficult to determine what sequences are important simply by looking at the genome of an organism. Instead, we look at fully processed strands of mRNA after transcription and splicing. The mRNA is isolated from cells and a DNA transcript complementary to that mRNA strand is created, called a cDNA. This cDNA is generated through the use of a protein called Reverse Transcriptase, a protein found in retroviruses that invade cells. The process is shown in the following diagram:
Thus, this process allows the generation of double stranded DNA that contains only the sequences that are translated into proteins. The ability to generate DNA that only contains useful sequences has several implications. It allows us to sequence the cDNA and then compare it to DNA sequences found in the genomic DNA. The location of the useful DNA sequences that encode proteins can be located this way, allowing geneticists to figure out the location of SNPs and mutations in organisms with genetic defects. In addition, it allows us to locate other important, but non-coding DNA sequences in the genome called Promoters and Enhancers. Promoters are sequences that RNA Polymerase binds to initiate transcription of a gene, while enhancers are sequences that proteins called Transcription Factors bind to and help to assemble the RNA Polymerase transcription complex to do this transcription. This is where Computational Genomics comes into play. After the sequencing of the genomes of different organisms, including humans, it was determined that although the genome of different organisms in the same species is over 99% identical, the 1% difference caused by SNPs usually have a pattern. Upon generation of cDNA transcripts and matching them to their location in the genome, it was determined that most polymorphisms exist in non-coding, often repeating sequences. It is hypothesized that the reason for this is because polymorphisms or mutations in useful sequences such as genes often produce deleterious alleles (alternative forms) of the gene, resulting in proteins that are defective and either lack its required function or become overly active, as in the case of cancers. Thus, organisms that acquire these mutations have diminished survival rates, and usually do not pass on the mutant genes to its progeny very well, thus getting the polymorphic and mutant genes out of the "gene pool". The results of genome sequencing projects are often stored into databases that geneticists retrieve to compare data with. As expected, the sequences that encode important proteins are highly conserved, with very little to no variation amongst different organisms and even different species. Computational genomics, however, also yielded a surprising result: in addition to the coding sequences being highly conserved, geneticists also noticed that some non-coding regions near the coding sequences were also highly conserved. These became known as Conserved Non-Coding Sequences (CNS) and were never identified until recently, when modern computing made it possible to compare the DNA sequences of a large number of organisms in a region of the genome to look for conserved sequences. These CNS were hypothesized to be the sequence for Promoters and Enhancers, which enable the gene to be transcribed. In addition, this hypothesis was further strengthened when organisms with polymorphisms in these regions showed deficiencies in the expression of that gene. With the advent of modern computing, it not only allowed geneticists to identify the location of genes, it also allowed the location of gene regulatory elements. Computational genomics is also used to analyze polymorphisms in the genome of different organisms of the same species. One application with the greatest potential is the use of computational genomics in the field of new experimental drugs. It's fair to say that most people have occasionally heard on the news about companies recalling new medications due to harmful effects on a few patients out of a large group. Geneticists in the field of computational genomics at the University of California, Berkeley attempt to answer the question of why does that small group of people in particular experience harmful effects from the medication while the rest seem to experience no side effects? To do this, they utilize gene chip technology. Gene chips are able to identify which allele is present in the genome of the organism by binding to specific sequences corresponding to the gene and fluorescing a different color, depending on which allele is bound to the host DNA. The color that is fluoresced is analyzed by computers and stored into a database. In the case of new experimental drugs, patients who suffer harmful effects from the drugs have their genome analyzed using gene chips. Computers then analyze the results of the gene chips from that group of patients and using specific algorithms, try to find genomic polymorphisms that are common amongst all the patients in that group, but are not found in patients that do not experience those side effects. If such a polymorphism is found, it can be hypothesized that people with the polymorphism will have a genetic inclination to suffer the harmful side effects. Furthermore, the gene with the polymorphism can be analyzed to determine what is the target protein that reacts with the drug and causes undesirable results. Finally, modern computing power allows for a very powerful tool to be utilized: networking and collaborative intelligence. One of the best examples of this is an ongoing project known as the Multilocus Sequence Typing (MLST) project. Rather than having one party collect genetic data about a specific organism, as is traditional in most fields, the MLST project utilizes the idea of collaborative intelligence to collect data for its database. The MLST project contains a database of alleles of housekeeping genes in strains of bacteria such as Bacillus Cereus, the bacteria responsible for most food poisoning cases out there. Laboratories that are researching such organisms sequence the genes of interest, and can identify them based on previous data stored in the MLST network database. Most of this data came from previous submissions of DNA sequences for the genes of interest. Thus, rather than having a specific party collect all the data, the network is made public so that any laboratories currently researching those organisms can submit their findings. From then on, laboratories that research the same organisms can compare their data to data collected by other labs and easily identify their findings. With networks such as MLST, laboratories can perform their research at a much quicker pace, as they are able to identify organisms based on previously submitted data from a collaborative effort, rather than having to start from scratch. This clearly shows the advantages that modern networks and databases provide for genomics research, making it cheaper, quicker, and much more efficient. |
||